min. read
Topics
Loss of OXR1 causes lysosomal dysfunction and a novel neurodevelopmental disorder
An international collaborative study has recently discovered a new neurodevelopmental disorder characterized by brain atrophy, speech and motor delays, hypotonia (low muscle tone), seizures and intellectual disability in five affected individuals with pathogenic variants in the Oxidative Resistance 1 (OXR1) gene. Studies in fruit flies unearthed a novel role for OXR1 in regulating the acidic environment of the lysosomes (“the cellular garbage recyclers”), disruption of which is likely the underlying cause of this disorder.
Image
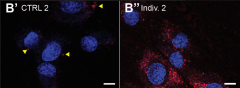
The study published in American Journal of Human Genetics and was jointly conducted in the laboratories of Dr. Hugo J. Bellen, professor at Baylor College of Medicine, investigator at the Howard Hughes Medical Institute and the Jan and Dan Duncan Neurological Research Institute, and Dr. Philippe Campeau, clinical associate professor at the University of Montreal.
“My lab had been studying the role of an epilepsy gene TBC1D24 in a neurological condition called the DOORS syndrome. To assess whether mutations in OXR1, a gene related to TBC1D24, also caused DOORS syndrome, we enrolled individuals with epilepsy and OXR1 mutations in our study. Clinical analysis revealed that OXR1 mutations were responsible for a new neurological syndrome, whose symptoms overlap with DOORS,” Campeau said. “With the help of the Undiagnosed Diseases Network team at Baylor, I connected with Dr. Bellen to further characterize this new disorder and to understand how OXR1 deficiency contributed to it. We combined our clinical data and cell studies with experiments in fruit flies to tease apart the mechanistic link between OXR1 mutations and severe neurological defects in the five patients.”
Lysosomal dysfunction is the cause for OXR1 deficiency associated symptoms
OXR1 is a complex gene with several transcripts that are expressed in many tissues during development. It was implicated in diverse and seemingly unrelated biological functions. However, one common function that appears to be conserved from microbes to mice is the ability of OXR1 to suppress oxidative and other forms of cellular stress. It remains unclear how the loss of its antioxidant action is linked to these diverse biological functions.
To understand how loss of OXR1 caused neurodevelopment symptoms in five patients, the team generated fruit flies that lacked mustard (mtd), the fly homolog of human OXR1 gene. Mutants lacking mtd specifically in the neurons died early and exhibited motor defects and seizures, similar to the affected individuals.
Interestingly, several experiments performed in cultured skin cells from affected individuals and in mtd flies showed that increased sensitivity to oxidative stress was not the primary driver of these symptoms.
“Detailed ultra-structural examination of mtd mutants revealed massive progressive accumulation of lysosomes and other trafficking intermediates with undigested contents - a clear indication that the lysosomes, which act as the cellular trash receptacles and recycling centers, were not functioning properly. Consistent with this, we also saw large numbers of lysosomes and aberrant lysosomal structures in the cultured skin cells of affected individuals, suggesting a lysosomal disorder,” Bellen said.
TLDc domain is sufficient to reverse mtd defects
Loss of mtd could be functionally replaced by the expression of a shorter version of OXR1 containing only the human TLDc region, a domain of unknown function. Surprisingly, expression of the TLDc domain of human Nuclear Receptor Coactivator (NCOA7), a protein that shares close structural homology to human OXR1 and fly mtd, was also sufficient to reverse defects in mtd mutant flies.
“Although OXR1 and NCOA7 are structurally similar, they were thought to have distinct non-overlapping functions – while deletion of OXR1 in rodents causes early death and severe loss of brain regions, mice lacking NCOA7 survive long and show kidney and urinary defects. However, our study shows the TLDc domain of human OXR1 and NCOA7 share a common function, suggesting that the two genes may act similarly at the molecular level,” said Julia Wang, graduate student in the Bellen lab.
“Our experimental detour into NCOA7 led us to molecular role for OXR1. Interestingly, both OXR1 and NCOA7 interact with V1-ATPase subunits, proteins that acidifies lysosomes. In fact, kidney cells of mice lacking NCOA7 had fewer V1-ATPAse levels and their urine was less acidic. Since lysosomes need to maintain an acidic environment for optimal degradation of unwanted proteins and other macromolecules, these data implicate lowered acidity of lysosomes as the underlying mechanistic cause for diverse symptoms associated with OXR1 deficiency – an exciting finding that opens avenues for further exploration into possible therapies for this novel disorder and other OXR1 deficiencies,” Bellen said.
Related Stories

News Release
Apr 10, 2026
Texas Children’s Receives $5 Million Estate Legacy Gift to Advance Pediatric Behavioral Health...

News Release
Apr 02, 2026
Houston Methodist and Texas Children’s Hospital start a collaborative program, backed by nearly...

News Release
Mar 30, 2026
Texas Children’s Austin & Ronald McDonald House Announce Opening of First In-Hospital House in ...

News Release
Mar 25, 2026
Texas Children’s Researcher Awarded Grant to Advance New Ways to Prevent and Treat Alzheimer’...

News Release
Mar 17, 2026
Golf Pros to Wear “Texas Children’s Cleats for Kids” at the Texas Children’s Houston Open...

News Release
Mar 12, 2026
Lurie Autism Institute Announces Dr. Huda Zoghbi as the Inaugural Recipient of the Nancy Lurie Ma...
20616
News Release
article