min. read
New study reveals potential drug targets for MECP2 duplication and Rett syndromes
Image
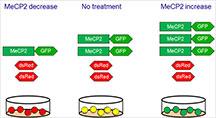
An exciting study published in Science Translational Medicinefrom the laboratory of Dr. Huda Zoghbi, director of the Jan and Dan Duncan Neurological Institute (NRI) at Texas Children’s Hospital, and professor at Baylor College of Medicine, describes the discovery of new ‘druggable’ modulators of methyl-CpG-binding protein 2 (MeCP2), a key neuronal protein. Too little MeCP2 protein causes Rett syndrome in girls and too much of it causes MECP2 duplication syndrome, a progressive neurological disorder in boys.
MeCP2 is one of the “Goldilocks” proteins which means minor changes in its level can lead to a variety of neurological symptoms including epilepsy, autism, cognitive dysfunction, and poor motor function in humans and mice.
Zoghbi’s team had previously shown that either deletion of an extra copy of the MECP2 gene or inhibition of production of excess MeCP2 protein using antisense oligonucleotides in mice modeling MECP2 duplication syndrome normalized MeCP2 levels and reversed most of the symptoms, surprisingly, even in mature mice that have been symptomatic for months. Other labs have shown that restoring MeCP2 in neurons successfully reversed symptoms of Rett syndrome in mice.
Despite successful manipulation of MeCP2 in animal models using genetic methods, until now, it had been difficult to develop a drug to directly and precisely modulate the levels of MeCP2 and little was known about its regulators.
To identify regulators of MeCP2, the authors employed an elegant strategy using a fluorescent cell-based assay and tested the ability of all known human kinases and phosphatases, crucial enzymes that activate or inhibit target proteins, to modulate MeCP2 levels.
Four candidate modulators (three kinases and a phosphatase) emerged from the cell-based screen, two of which were further validated in mice. To investigate the potential relevance to MECP2 duplication syndrome, Zoghbi’s team tested the effect of inhibiting these candidate modulators in transgenic mice that express twice the normal levels of MeCP2 due to the addition of a copy of the human MECP2 gene. Only one candidate modulator had commercially available inhibitors, which when administered to those transgenic mice, reduced MeCP2 levels in the brain and improved their motor function.
Unintended side effects or cellular toxicity are the main reasons why many potential drug targets fail to become successful therapies and it takes decades of further preclinical research to identify new targets. The strategy described in this study can now be expanded to screen thousands of druggable targets to identify those that either decrease or increase MECP2 (to help solve MECP2 duplication syndrome and Rett syndrome, respectively). Having a rich source of targets will allow researchers to select the most efficacious candidate(s) with the least side effects and will greatly speed up the process of finding a treatment for MeCP2 disorders.
In addition to the promise it offers for MeCP2 disorders, this study also provides a blueprint to unearth potential drug targets for other neurological disorders caused by changes in expression of a specific protein.
Related Stories





20676
Media
article
